Revista Veterinaria. 2026; 37(1)
https://doi.org/10.30972/vet.3719224
Artículo Original


J.A Titi Pacosoncco1
H Vilcanqui Mamani1
F Vilca de Díaz1
Resumen
El objetivo de este estudio fue evaluar la diversidad genética y la estructura poblacional del gen mitocondrial citocromo b (cytb) en ganado Brown Swiss criado en el altiplano peruano. Se recolectaron muestras sanguíneas de 41 vacas procedentes de 13 establos lecheros en la provincia de Azángaro, Puno. Tras la extracción de ADN y amplificación por PCR, se realizó secuenciación Sanger; previo al análisis bioinformático, se excluyeron tres lecturas por baja calidad (Q-score < 20), trabajando con un conjunto final de 38 secuencias. El alineamiento se realizó con el algoritmo MUSCLE y la reconstrucción filogenética mediante Máxima Verosimilitud (modelo HKY+I) en MEGA v.12, mientras que los índices de diversidad se calcularon en R. Se identificaron 31 haplotipos distintos, revelando una alta diversidad haplotípica (Hd = 0,9687) en contraste con una baja diversidad nucleotídica (π = 0,0240). La red de haplotipos exhibió una topología en estrella, dominada por haplotipos únicos y pocos centrales. Asimismo, el Test de Neutralidad de Tajima arrojó valores negativos consistentes (D < -2,5), lo que sugiere un exceso de polimorfismos de baja frecuencia, indicativo de una expansión poblacional reciente o selección purificadora. El análisis filogenético mostró múltiples linajes divergentes sin una estructura geográfica definida. Se concluye que, a pesar de ser una raza comercial, la población de Brown Swiss en el norte de Puno mantiene una reserva genética mitocondrial considerable y heterogénea, probablemente debida al flujo genético activo, lo cual es fundamental para diseñar estrategias de conservación y mejora genética sostenible en la región.
Palabras clave: ADN mitocondrial, haplotipos, filogenia, distancia, linaje.
Abstract. The aim of this study was to evaluate the genetic diversity and population structure of the mitochondrial cytochrome b (cytb) gene in Brown Swiss cattle raised in the Peruvian highlands. Blood samples were collected from 41 cows belonging to 13 dairy farms in Azángaro province, Puno Region. After DNA extraction and PCR amplification, Sanger sequencing was performed. Prior to bioinformatics analysis, three sequences were excluded due to low quality (Q-score < 20), resulting in a final dataset of 38 sequences. Alignment was conducted using the MUSCLE algorithm, and phylogenetic reconstruction was performed using the Maximum Likelihood method (HKY+I model) in MEGA v.12. Genetic diversity indices were estimated using R. A total of 31 distinct haplotypes were identified, indicating high haplotype diversity (Hd = 0.9687) combined with low nucleotide diversity (π = 0.0240). The haplotype network exhibited a star-like topology, characterized by a high number of unique haplotypes and a few central ones. Tajima's Neutrality Test yielded consistently negative values (D < -2.5), suggesting an excess of low-frequency polymorphisms, which is consistent with a recent population expansion or purifying selection. Phylogenetic analysis revealed the presence of multiple divergent mitochondrial lineages without a clear geographic structure. These results indicate that, despite being a commercial breed, the Brown Swiss population in northern Puno retains considerable and heterogeneous mitochondrial genetic reserve, likely maintained through active gene flow. This genetic variability represents an important resource for the design of sustainable conservation and genetic improvement strategies in the region.
Key words: mitochondrial DNA, haplotypes, phylogeny, distance, lineage.
El ganado Brown Swiss aporta significativamente a la producción pecuaria por su robustez productiva, resistencia a enfermedades y adaptabilidad ambiental (Davidescu et al. 2024, Cartuche-Macas et al. 2025). Sin embargo, su uso extensivo plantea interrogantes sobre la posible reducción de diversidad genética en poblaciones receptoras (Wirth et al. 2021). En el contexto del altiplano peruano, y específicamente en la región de Puno, esta raza se ha consolidado como predominante; no obstante, existe un vacío de información sobre la estructura genética molecular de estos rebaños locales, lo que impide determinar si las prácticas actuales de manejo están afectando su variabilidad y potencial adaptativo.
Para abordar esta problemática, el análisis del ácido desoxirribonucleico (ADN) mitocondrial, específicamente el gen citocromo b (cytb), constituye una herramienta valiosa para estudios evolutivos debido a su herencia materna, ausencia de recombinación y tasa de mutación que va de moderada a alta (Demir et al. 2023). La diversidad haplotípica y nucleotídica del gen cytb refleja la variación genética en el linaje materno, con implicaciones para rasgos productivos y adaptativos (Dorji et al. 2022). Se considera que una alta diversidad indica mayor reserva genética favorable para la adaptación, mientras que valores bajos sugieren endogamia o cuellos de botella poblacionales (Cartuche-Macas et al. 2025).
Asimismo, los análisis filogenéticos basados en secuencias del gen cytb permiten esclarecer relaciones evolutivas y estructura poblacional (Gray 1989), información esencial para estrategias de selección y conservación sostenible. Adicionalmente, estos análisis pueden revelar evidencias de selección natural o artificial sobre este gen (Signer-Hasler et al. 2017), proporcionando datos sobre adaptación a diversas condiciones ambientales (Terefe et al. 2023).
Considerando la importancia de estos parámetros para la gestión ganadera regional, el objetivo de este estudio fue evaluar la diversidad genética y estructura poblacional del gen mitocondrial citocromo b (cytb) en vacunos Brown Swiss de establos lecheros en el norte de Puno, Perú.
Lugar de estudio y animales. El estudio se llevó a cabo en 13 establos lecheros ubicados en el distrito de Asillo de la provincia de Azángaro del departamento de Puno -Perú; localizado a una altitud media de 3,907 m.s.n.m. cuya geolocalización se encuentra en una latitud sur de -14.784097, y longitud oeste de -70.357005. Para el presente estudio se tomaron muestras de 41 vacunos (hembra) de la raza Brown Swiss criadas bajo un sistema semi-intensivo. El muestreo realizado a los animales fue aleatorio en cada hato lechero.
Toma de muestras, extracción de ADN y secuenciación. El estudio contó con la aprobación del comité de ética institucional y el consentimiento de los propietarios, cumpliendo con la normativa de bienestar animal vigente (Ley N° 30.407). Bajo estrictas medidas de bioseguridad y uso de material estéril, se colectaron muestras de sangre (6 ml) de 41 animales mediante venopunción de la vena caudal, previa desinfección de la zona con alcohol yodado al 3%. La obtención de ADN se inició con la separación de células nucleadas mediante centrifugación en gradiente de Ficoll. Seguidamente, la lisis celular y digestión proteica se efectuaron con buffer específico y proteinasa K a 37 °C. El ADN se purificó mediante extracción con fenol-cloroformo-alcohol isoamílico, seguido de tratamiento con RNAsa para eliminar ARN. Finalmente, la precipitación con isopropanol frío y lavados con etanol al 70% aseguraron la pureza del ADN, cuya integridad se verificó por electroforesis en gel de agarosa. A continuación, se amplificó un fragmento de 922 pb del gen citocromo b, correspondiente a las coordenadas 14.399 a 15.320 del genoma mitocondrial de referencia de Bos taurus (GenBank Acc. No. NC_006853). Esta región abarca el segmento central y el extremo 3' del gen. La amplificación se realizó mediante PCR convencional con un perfil térmico de desnaturalización inicial a 95 °C, seguido de 35 ciclos de desnaturalización a 95 °C, alineamiento a 55 °C y extensión a 72 °C, finalizando con una extensión final a 72 °C durante 10 minutos. Posteriormente, la secuenciación se realizó mediante el método de Sanger, empleando la química BigDye Terminator v3.1 en un secuenciador automático ABI 3130xl.
Alineamiento de secuencias. El análisis partió de un total de 41 muestras recolectadas. Previo al procesamiento, se realizó un control de calidad donde se excluyeron tres secuencias por no alcanzar un puntaje de calidad mínimo (Q-score) ≥ 20. En consecuencia, el alineamiento múltiple definitivo se llevó a cabo con las 38 secuencias restantes del gen cytb. Para este proceso se utilizó el algoritmo MUSCLE ejecutado en el software de análisis evolutivo MEGA v.12.0.10 (Kumar et al. 2024). La configuración incluyó parámetros estrictos, tales como una penalización de apertura de gaps de -200.00 y una penalización de extensión de 0.00. El algoritmo operó inicialmente mediante el método UPGMA en las primeras iteraciones, cambiando posteriormente a Neighbor Joining para refinar la precisión filogenética, estableciéndose una longitud mínima de diagonal (Lambda) de 20 para optimizar la identificación de regiones homólogas.
Selección de modelo y reconstrucción del árbol filogenético del gen cytb. El mejor modelo evolutivo fue seleccionado utilizando el programa MEGA (Kumar et al. 2024), mediante el criterio de máxima verosimilitud, evaluando 24 modelos de sustitución nucleotídica según sus valores de verosimilitud, BIC y AIC (Criterio de información Bayesiano y Criterio de información de Akaike respectivamente). El modelo HKY (Hasegawa-Kishino-Yano) con sitios invariantes (HKY+I) fue el que presentó el mejor ajuste. La reconstrucción filogenética se realizó bajo el mismo criterio, empleando el algoritmo heurístico Nearest-Neighbor Interchange (NNI) y evaluando la confiabilidad de los clados mediante 1.000 réplicas de bootstrap. Se utilizó la opción "Use all sites" sin aplicar filtros de ramificación, optimizando el rendimiento con paralelización en tres hilos.
Estimación de Distancia Genética en cytb. El análisis se llevó a cabo en MEGA (Kumar et al. 2024) y se basó en el promedio global (Overall Mean), con 1.000 réplicas de bootstrap para calcular la varianza e intervalos de confianza. Se empleó el modelo de Máxima Verosimilitud Compuesta, considerando transiciones y transversiones, bajo supuestos de tasas de sustitución uniformes y evolución homogénea entre linajes. Para evaluar la robustez de las estimaciones, se aplicaron tres métodos de manejo de datos faltantes: deleción por pares, deleción completa y deleción parcial.
Análisis Estadístico. Para determinar las divergencias genéticas en el gen cytb, se utilizó el Test de Neutralidad de Tajima (D), que compara la diversidad genética promedio por pares (π) con la esperada bajo neutralidad basada en sitios segregantes (S); un valor positivo sugiere selección balanceadora o reducción poblacional, mientras que un valor negativo indica selección purificadora o expansión poblacional (Tajima 1989). Donde S representa (número de sitios polimórficos), π (diversidad nucleotídica), θ (estimación de diversidad basada en S) y D (explica la varianza) (Nei y Kumar 2000). El análisis se llevó a cabo en el programa de análisis genético de evolución molecular MEGA v.12.0.10 (Kumar et al. 2024). Para estimar el número de haplotipos y la diversidad haplotípica (Hd), se utilizó el programa estadístico R v.4.5.0 utilizando las librerías “ape” (Paradis et al. 2024) y “pegas” (Paradis et al. 2023) con los parámetros predeterminados de cada librería.
Test de Neutralidad de Tajima (D). Los resultados del test de neutralidad (Tabla 1), revelaron valores negativos y consistentes del estadístico D de Tajima en los tres métodos de deleción: -2,65944 (completa), -2,89106 (por pares) y -2,85358 (parcial), indicando un exceso de polimorfismos de baja frecuencia. La diversidad nucleotídica (π) varió entre 0,01027 y 0,02203, mientras que los valores de θ oscilaron entre 0,06794 y 0,08391. Esta discrepancia sustenta la señal de desviación respecto a la neutralidad. El número de sitios segregantes (S) fue variable (55 a 192), reflejando diferencias en la inclusión de regiones polimórficas según el tratamiento de datos faltantes. En conjunto, los resultados sugieren la acción de selección purificadora o una expansión poblacional reciente, escenarios esperables en genes mitocondriales funcionales como cytb, sujeto a fuerte presión evolutiva.
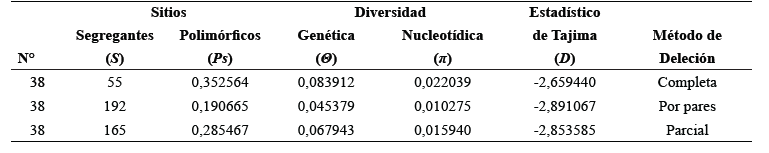
Estos resultados coinciden con estudios previos realizado por Ratnam et al. (2022) en ganado autóctono (D = -2,40) y son más pronunciados que los reportados por Owaid et al. (2020) para ganado Holstein iraquíes (D = -1,67053), apoyando la hipótesis de expansión poblacional reciente en razas comerciales. En contraste, los valores positivos observados en razas chinas (Yan et al. 2019) y ganado cruzado iraquí (Owaid et al. 2020) reflejarían historias demográficas distintas, con posibles señales de selección balanceada o contracción poblacional.
La consistencia de nuestros hallazgos entre los tres métodos de procesamiento de gaps, junto con estudios previos sobre alta variabilidad intra-poblacional (Mkize y Zishiri 2020), sugiere que las razas comerciales presentan dinámicas evolutivas particulares, posiblemente asociadas a su manejo reproductivo intensivo y flujo génico globalizado. Estos resultados proporcionan información clave sobre los procesos microevolutivos que moldean la diversidad genética en poblaciones bovinas bajo distintos regímenes de selección.
Diversidad Haplotípica (Hd) y nucleotídica (π). El análisis de las secuencias permitió la identificación de 31 haplotipos distintos. Se obtuvo una diversidad haplotípica (Hd) de 0,9687 y una diversidad nucleotídica (π) de 0,0240; estimadas mediante el método de deleción por pares.
Con respecto a la genealogía, la red de haplotipos (Figura 3) exhibió una topología en forma de estrella, caracterizada por dos haplotipos centrales (H1 y H2) y múltiples haplotipos derivados en posición periférica, conectados principalmente por eventos de mutación puntual. El haplotipo H3 se identificó como el de mayor frecuencia, mientras que la mayoría de las variantes restantes correspondieron a haplotipos únicos o de baja frecuencia.
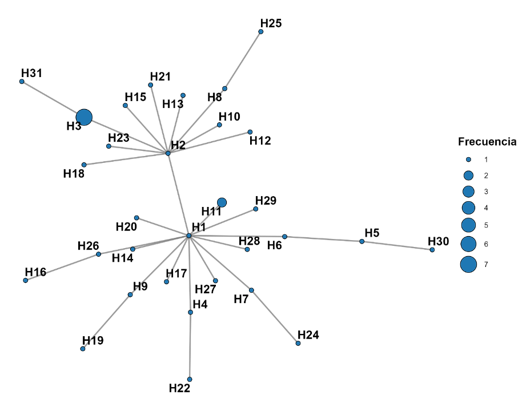
Los índices obtenidos revelan una elevada heterogeneidad haplotípica coexistiendo con diferencias nucleotídicas bajas, un patrón genético característico que sugiere un evento de expansión demográfica reciente. La diversidad reportada en este estudio (Hd = 0,9687) supera ampliamente los valores descritos por Valizadeh et al. (2011) en ganado iraní (π = 0,0037) y por Hartatik et al. (2019) en ganado indonesio (Hd = 0,3778; π = 0,00099). Del mismo modo, contrasta notablemente con la diversidad moderada observada en ganado nativo de Indonesia (Hd = 0,515; Prihandini et al. 2020), Etiopía (Hd = 0,7540; Tarekegn et al. 2018) y Corea (Chung 2013). Estas discrepancias podrían atribuirse a diferencias en el tamaño efectivo de la población y a las distintas estrategias reproductivas o historias demográficas de cada región.
Es relevante destacar que la alta proporción de haplotipos únicos encontrada coincide con lo reportado específicamente para la raza Brown Swiss por Hansen (1997), quien describió una incidencia del 89% de haplotipos únicos, diferenciándose de otras razas bovinas. Por otro lado, la distribución de los haplogrupos y la topología de estrella observada concuerdan parcialmente con estudios en ganado japonés y coreano (Sasazaki et al. 2006), sugiriendo la posible coexistencia de tres linajes maternos diferenciados y la conservación de linajes ancestrales dentro de la raza.
En síntesis, la alta variabilidad haplotípica detectada en el ganado Brown Swiss de la zona norte de Puno resalta un acervo genético valioso. A pesar de la influencia de factores como la selección artificial y la domesticación, esta población mantiene una diversidad que favorece su adaptabilidad futura, subrayando su importancia para la implementación de programas de conservación y mejoramiento genético regional.
Distancia Genética en cytb. La estimación de la distancia genética media entre las 38 secuencias del gen cytb en vacunos Brown Swiss mostró valores congruentes entre métodos de tratamiento de gaps, aunque con variaciones en la precisión (Tabla 2). La deleción parcial arrojó una distancia de 0,0311 ± 0,1074, mientras que la deleción completa presentó el valor más alto 0,0357 ± 0,4701 pero con una alta incertidumbre. En contraste, la deleción por pares mostró la menor distancia genética 0,0241 ± 0,0169 y el error estándar más bajo, lo que sugiere una estimación más precisa al aprovechar la máxima cantidad de datos comparables entre pares de secuencias.
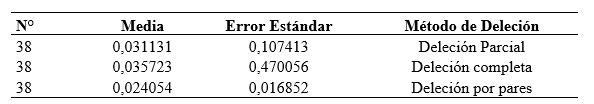
Estos resultados coinciden con la baja diversidad nucleotídica y los valores negativos del Test de Tajima, reforzando la evidencia de una diversidad genética moderada a baja en esta población, posiblemente influida por eventos recientes de expansión o selección.
Estos valores son inferiores a los reportados por Prihandini et al. (2020) en ganado Bali (0,092), lo que sugiere mayor homogeneidad genética en Brown Swiss, posiblemente debido a selección artificial intensiva. En contraste, estudios en ganado indonesio realizado por Rahim et al. (2021) muestran mínima divergencia con Bos javanicus (0,000 - 0,001), reflejando su cercanía evolutiva. Nuestros resultados también coinciden con los hallazgos de Hansen et al. (2011), que indican un origen materno común entre razas comerciales (Holstein, Jersey, Brown Swiss) a pesar de su diferenciación genómica.
La variabilidad en cytb tiene también implicaciones prácticas en otros campos, por ejemplo, en la detección de adulteración en lácteos (Mininni et al. 2009) y en estudios filogenéticos (Pal et al. 2019).
Árbol Filogenético. El árbol filogenético reconstruido mediante máxima verosimilitud con el modelo HKY+I (Figura 4) revela una estructura poblacional con baja diferenciación genética entre las 38 secuencias del gen cytb en vacunos Brown Swiss. La topología presenta una disposición en peine, característica de poblaciones con expansión reciente, y evidencia múltiples linajes que divergen desde nodos basales sin una clara estructura jerárquica.
El soporte estadístico, evaluado mediante 1.000 réplicas de bootstrap, fue en su mayoría bajo o nulo, con algunos clados de soporte moderado, por ejemplo, los animales identificados como vaca29 con vaca31 (65%) y vaca18 con vaca21 (39%). Un clado superior, más diversificado (bootstrap 58%), agrupa principalmente a las secuencias de vaca39, vaca18 y vaca21. Esta configuración filogenética es coherente con los bajos valores de distancia genética y los resultados del Test de Tajima, lo que sugiere una población homogénea, sujeta a expansión demográfica reciente o selección purificadora, donde predominan mutaciones recientes no fijadas.

El análisis del gen mitocondrial cytb en ganado Brown Swiss del norte de Puno en Perú, demostró alta diversidad genética materna, evidenciada por 31 haplotipos y una diversidad haplotípica de 0,9687. Los resultados sugieren expansión poblacional reciente y flujo génico activo entre establos, manteniendo homogeneidad estructural poblacional. Esta diversidad mitocondrial constituye un recurso genético valioso que sustenta el desarrollo de programas de conservación y mejoramiento genético orientados a optimizar la productividad mientras se preserva la variabilidad genética en esta población altoandina.
Agradecimiento. Los autores agradecen el financiamiento realizado por la Federación de Docentes Universitarios (FEDU) de la Universidad Nacional del Altiplano Puno - Perú.
Contribución de los autores. TPJA: Conceptualización, Metodología, Investigación, Curación de datos, Análisis formal, Adquisición de financiamiento, Administración del proyecto, Redacción – borrador original, Redacción – revisión y edición. MVH: Investigación, Análisis formal, Validación, Redacción – revisión y edición. VDF: Investigación, Análisis formal, Redacción – revisión y edición.
Declaración de conflictos de intereses. Los autores declaran no tener conflictos de intereses financieros ni relaciones personales conocidas que pudieran haber influido en el trabajo presentado en este artículo.
Disponibilidad de datos. Los datos estarán disponibles previa solicitud.
